文章来源:蒲公英Ouryao
2025年5月6日,美国FDA宣布,将扩大对美国海外药品生产厂进行无预先通知的这一“突袭”式检查方式,涵盖食品、关键药品等出口美国的医疗产品。
FDA局长马卡里(Marty Makary)表示,美国海外生产商长期以来享有“双重标准”——在检查前得到提前通知,而美国本土生产商则无任何预警:“这一格局今天终结”。
FDA新闻稿强调,此举的目的是让美国海外企业与国内企业接受同等严格的监管,并依托此前在印度和中国试点的无预告检查项目,全面提升海外监管力度。
这一监管调整与特朗普总统于2025年5月签署的行政令密切相关。
特朗普5月5日的行政令指出,美国国内药厂设施建设面临FDA突击检查等多重监管壁垒,使新设施建设耗时5至10年难以接受;相比之下,海外生产商检查频次更低。为恢复美国药品供应链安全,行政令要求FDA在90天内制定方案,完善对向美国供货的海外药厂的风险评估和检查制度,包括在法律允许范围内对美国海外生产商提高检查费率,并向公众披露各国年度检查数量。
换言之,新行政令不仅致力于精简美国国内审查流程,更明确要加大对海外设施的现场监管和透明度。
早在2022年底通过的2023财年拨款法案中,就已包含相关安排。该法案第3615条规定设立试点项目,要求FDA“增加对美国海外药品生产企业的突袭式监督检查”,并评估其与美国国内检查的差异。此举意味着,国会层面也认识到长期存在的监管不平衡,鼓励FDA试水“突袭式”海外检查。2024年美国国会对FDA海外监管的质疑不断。
能源商务委员会主席卡西·罗杰斯(Cathy McMorris Rodgers)在听证会上批评FDA:“海外检查本就一直名存实亡,在疫情之前就存在问题,疫情期间几乎停摆,到现在进展也很有限”。她指出,“实际上我们对国内企业要求要严格得多,对海外企业却放宽了……我们需要一个鼓励国内生产的公平竞争环境”。美国政府问责局(GAO)也多次报告指出,由于资源有限,FDA过往往会提前几周通知海外检查对象,使对方有时间临时“打扫门面”,难以见到设施的日常真实状况。
综合来看,此次政策调整是一系列因素共同作用的结果。
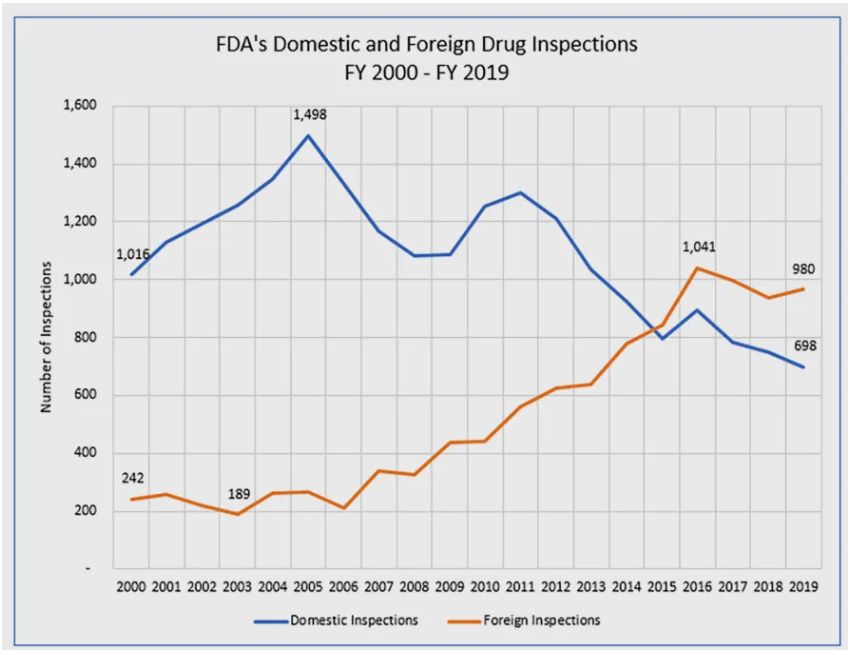
药品全球化趋势下,美国对外依赖程度日益提高:GAO报告显示,超过50%的美国药品生产由海外企业承担,尤其是中国和印度生产商占据了关键原料供应的主导地位。疫情以来,FDA海外检查严重滞后。分析发现,近两千家应巡厂曾逾期未接受例行检查。国会授权、拨款支持及行业安全事件叠加发力,共同推动了监管方向调整。
2023年国会明确授权,突袭式检查试点项目开始实施,并要求持续评估效果。2024年多起药品质量事故也引发担忧:例如,2023年印度阿默达巴德市INTAS公司生产的眼药水被检出携带超耐药细菌,引发80余名美国患者感染,其中4人死亡。这些事件凸显了对海外生产安全的长期隐忧,也加大了美国加强海外监管的社会和政治压力。
特朗普行政令和新拨款法案的逻辑是一致的:通过加快国内生产商审批和供应链本土化减轻对外依赖,同时对外资深度审查以保障质量。这一政策演进可视为美国应对全球化挑战的综合策略。在此背景下,FDA当前的改革方向——把美国国内常用的突击检查模式推广到海外——正是其长期积累问题的直接回应。
美国监管专家普遍认为,更严格的海外监管对保障药品安全至关重要。杜克大学教授大卫·里德利(David Ridley)警告:“仿制药厂面临降本压力,一些生产商会牺牲质量。如果不检查,我们只有在悲剧发生后才知道问题”。美国FDA助理专员迈克尔·罗杰斯(Michael Rogers)则强调,FDA全球监管检查是确保进入美国市场产品“安全、可靠”的“金标准”。FDA局长马卡里也表示,面对有限的人力资源,机构将缩短每次检查时间,实现“更快进入、更快退出”,从而用相同人员完成更多检查。这表明监管层既认识到加强检查的重要性,也在寻求提高效率以克服人力不足的问题。
与此同时,也有声音提醒可能的挑战。一些行业分析认为,大规模海外突击检查需要投入大量资源:FDA海外办事处和检查员数量有限,地缘与语言文化差异也可能影响检查效率。另外,此举可能引发国际摩擦——例如中印两国若认为检查过于严苛,可能会进行外交交涉。不过FDA及白宫均表示,此次行动是落实长期监管策略、无意针对特定国家,其核心目的是从根本上提升全球药品质量和供应链安全。
根据美国《联邦食品、药品与化妆品法案》,任何向美国市场供应药品的工厂都必须在FDA注册并接受监管。
FDA的检查主要分为三类:上市前检查(pre-approval)、上市后常规监督检查(surveillance)、以及针对出现安全或质量问题时的有因检查(for-cause)。
在常规监督检查中,FDA采用风险评估模式(Site Selection Model)优先审查潜在风险最高的生产商。传统上,美国境内的检查通常是突击进行,只有特殊项目才会预先通知;海外检查过去多以预约方式进行。
新政意味着,FDA将把国内的检查模式移植到海外。也就是说,未来对许多海外生产商的例行监督检查将无需预先通报。按照FDA规定,若检验中发现严重违规,可通过下达进口警示(禁止该厂产品进入美国)或召回产品来保护患者。
此外,2012年生效的法案明确:任何拖延、拒绝接受FDA检查的美国海外生产商,其所有药品都将被视为“掺假”,无法合法进口。FDA强调,所有检查都严格以科学标准进行,无论是国内还是海外,都需符合同样的质量规范。此次政策调整等同于强调全球药品审查的无缝衔接——让每一件进入美国家庭的药品,都经过同等严谨的审查程序。
新政实施后,全球医药供应链格局可能发生变化。对中国和印度这样的主要药品生产国而言,FDA的监管力度升级无疑带来更严格的审查要求。这两个国家生产了美国市场所需大量仿制药原料,美国对其依赖度极高。
数据显示,超过半数的美国药品生产外包给海外厂家,国会听证会指出中印合计制造了接近70%的美国仿制药活性成分(API)。因此,中印药企将成为首当其冲的检查对象。
以印度为例,INTAS制药厂事件已引发美国监管层高度关注。FDA在宣布新政时也明确提到印度和中国已有无预告检查试点,未来预期更多类似行动。
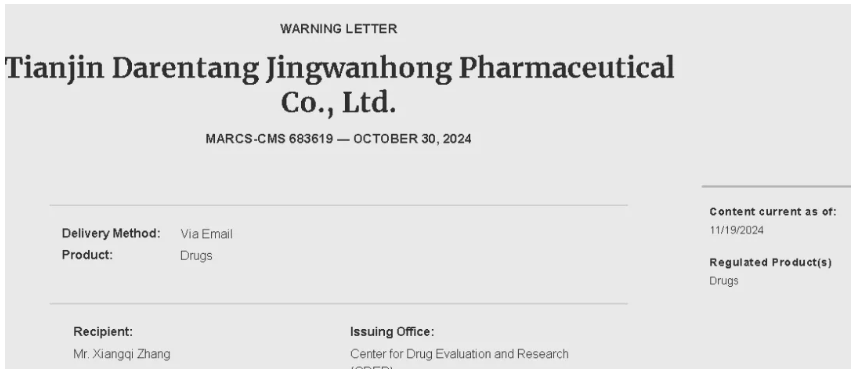
对中国而言,监管行动和案例日益增多:2024年,FDA曾对天津市一家中成药生产企业下发警告函,称该厂限制并拒绝接受FDA检查,其生产的药品依法被认定为“掺假”;2025年2月,FDA再次对四川一家API制造企业提出警告,指出其在原料检测等质量管理方面存在严重偏差。
可以预见,新政的落实可能促使中印生产商提高质量合规投入,以应对更频繁且无预警的检查。反过来,这可能使部分低成本供应商的竞争优势受到挑战,潜在导致药品成本上升或供应重组。
参考资料:FDA官网新闻稿,美国白宫行政令,2023年联邦拨款法案,媒体报道及专业分析等。https://www.fda.gov/news-events/press-announcements/fda-announces-expanded-use-unannounced-inspections-foreign-manufacturing-facilities?utm_medium=email&utm_source=govdeliveryhttps://www.whitehouse.gov/presidential-actions/2025/05/regulatory-relief-to-promote-domestic-production-of-critical-medicines/https://www.whitehouse.gov/fact-sheets/2025/05/fact-sheet-president-donald-j-trump-announces-actions-to-reduce-regulatory-barriers-to-domestic-pharmaceutical-manufacturing/#:https://www.raps.org:text=The%20Order%20directs%20the%20U.S.,provide%20early%20support%20before%20facilitieshttps://www.raps.org/news-and-articles/news-articles/2025/5/fda-to-expand-unannounced-foreign-inspections-afte